2.F: STRING & rbioapi
Moosa Rezwani
2026-07-30
Source:vignettes/rbioapi_string.Rmd
rbioapi_string.RmdIntroduction
STRING is a comprehensive database of known and predicted protein-protein associations. It integrates direct physical interactions and indirect functional associations from experimental evidence, curated databases, text mining, co-expression, and computational predictions. rbioapi provides a consistent R interface to STRING’s mapping, network, homology, annotation, and enrichment API resources.
The species argument
Most rbioapi STRING functions accept a species argument
containing an NCBI
Taxonomy identifier, such as 9606 for human. Supplying it is
recommended because it helps STRING resolve ambiguous identifiers.
For the network, interaction-partner, homology, PPI-enrichment,
network-image, and enrichment-image functions, species is
required when the input contains more than 10 unique IDs. Mapping,
functional-enrichment, and functional-annotation requests do not have
this local restriction. The limit is based on unique IDs rather than the
length of the original vector.
Map your IDs to STRING IDs
Although STRING recognizes a variety of identifiers, mapping them to STRING IDs before using other rbioapi STRING functions makes subsequent requests faster and less ambiguous.
## 1. Create a vector of protein identifiers
proteins <- c(
"p53", "BRCA1", "cdk2", "Q99835", "CDC42", "CDK1", "KIF23",
"PLK1", "RAC2", "RACGAP1", "RHOA", "RHOB", "PHF14", "RBM3"
)
## 2. Map the protein identifiers
proteins_mapped_df <- rba_string_map_ids(ids = proteins, species = 9606)
## 3. Use the mapped STRING IDs in the remaining examples
proteins_mapped <- proteins_mapped_df$stringIdGet interaction network of a protein set
You can retrieve interactions among the proteins in your set,
together with the scores for each STRING evidence channel. Use
required_score to set the minimum interaction score and
network_type to select functional or physical
associations.
See the Value section of the
rba_string_interactions_network() manual for descriptions
of the returned columns.
int_net <- rba_string_interactions_network(
ids = proteins_mapped,
species = 9606,
required_score = 500
)Instead of supplying protein identifiers, you can retrieve the
network formed by proteins annotated with a STRING functional term. In
this mode, set ids = NULL and supply both
network_term_id and species.
rba_string_functional_terms() can search for functional
terms using either an identifier or descriptive text:
tcr_terms <- rba_string_functional_terms(
term_text = "T cell receptor signaling pathway",
species = 9606
)The first match for this search is GO:0050852, which
identifies the GO term “T cell receptor signaling pathway”:
term_net <- rba_string_interactions_network(
ids = NULL,
network_term_id = "GO:0050852",
species = 9606,
required_score = 900
)The proteinCount column contains the complete number of
proteins annotated with each term, while preferredNames and
stringIds are returned as list-columns. The
ids and network_term_id input modes cannot be
combined.
Get interaction partners of a protein set
The previous example returned interactions among the input proteins.
To retrieve proteins that interact with one or more input proteins,
including proteins outside the input set, use
rba_string_interaction_partners():
## This example uses one protein (CD40), but `ids` can contain multiple proteins
int_partners <- rba_string_interaction_partners(
ids = "9606.ENSP00000361359",
species = 9606,
required_score = 900
)Get network image of a protein set
STRING can render the interaction network as a static image. The
rba_string_network_image() function supports several
options for controlling the network contents and appearance; see the
function manual for details. It also supports the same
network_term_id input mode described above.
## Example 1:
graph_ppi1 <- rba_string_network_image(
ids = proteins_mapped,
image_format = "image",
species = 9606,
save_image = FALSE,
required_score = 500,
network_flavor = "confidence"
)For example, the network for a functional term can be rendered with:
term_image <- rba_string_network_image(
ids = NULL,
network_term_id = "GO:0050852",
species = 9606,
required_score = 900,
save_image = FALSE
)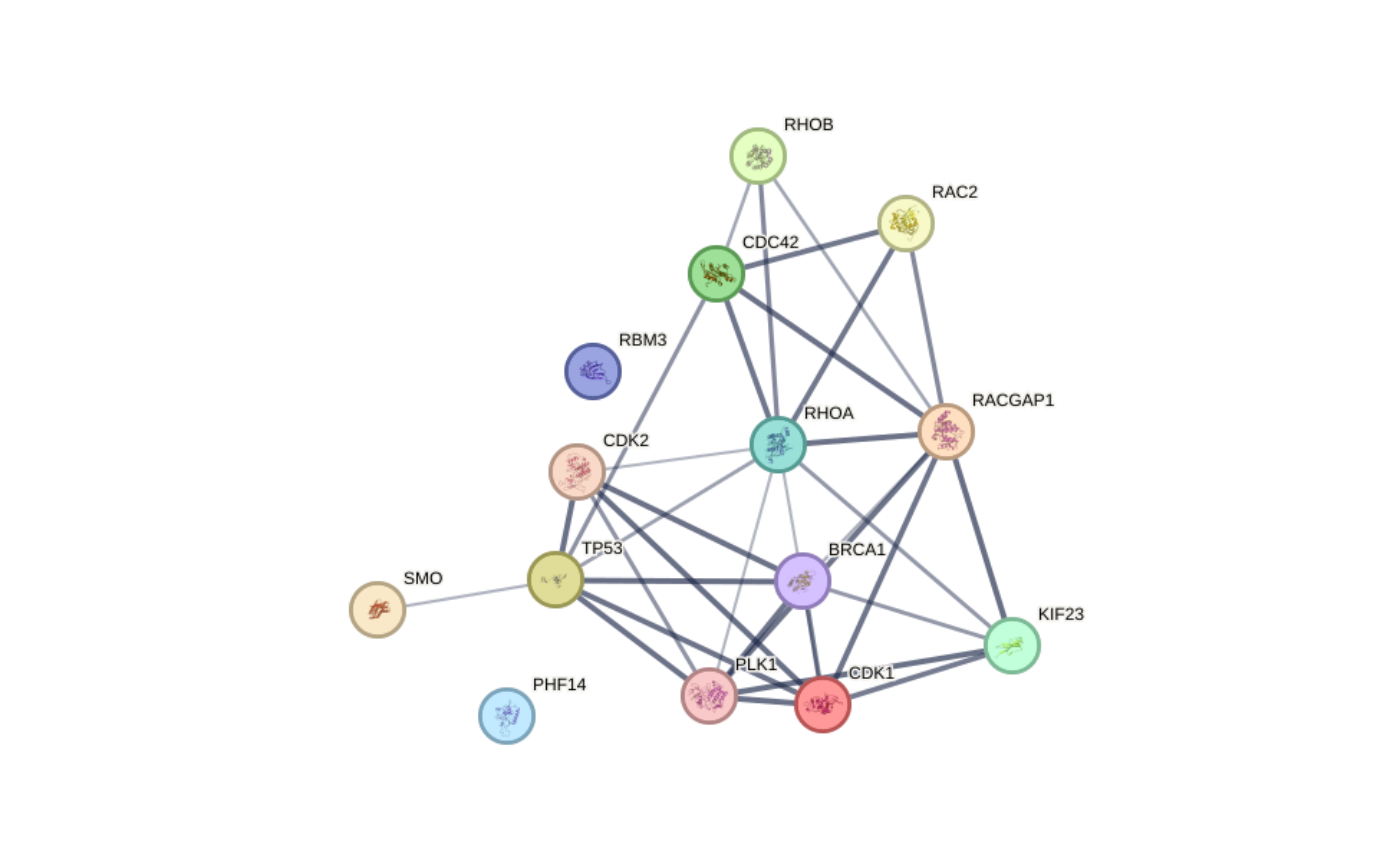
Network images - Example 1
## Example 2:
graph_ppi2 <- rba_string_network_image(
ids = proteins_mapped,
image_format = "image",
species = 9606,
save_image = FALSE,
required_score = 500,
add_color_nodes = 5,
add_white_nodes = 5,
network_flavor = "actions"
)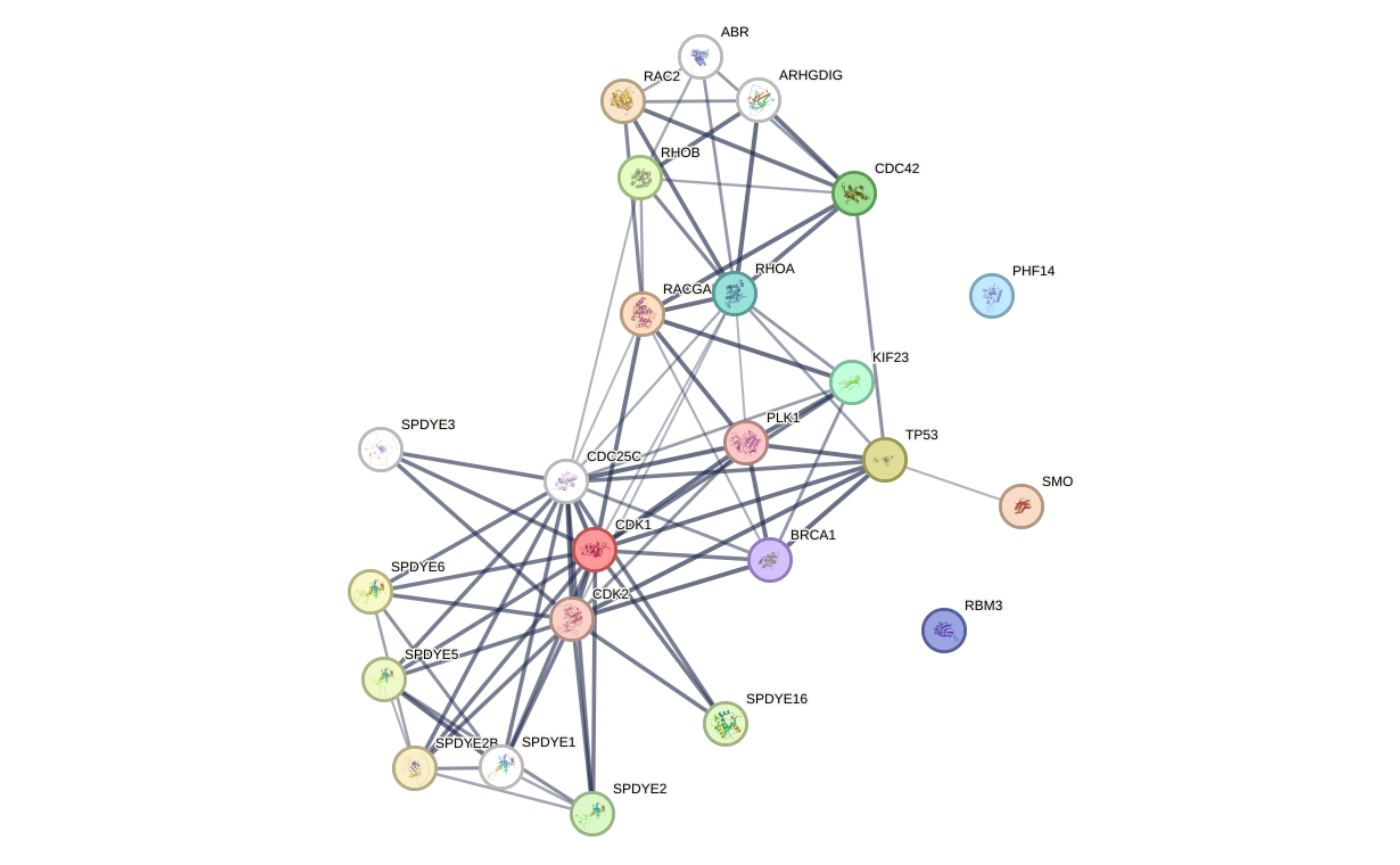
Network images - Example 2
Enrichment analysis using STRING
STRING supports two complementary enrichment analyses: functional enrichment and protein-protein interaction enrichment. See the STRING publication for methodological details.
Functional enrichment
Functional enrichment tests the supplied protein set against annotation resources including Gene Ontology, KEGG pathways, UniProt Keywords, PubMed publications, Pfam domains, InterPro domains, and SMART domains. The results include raw p-values and false discovery rates. See the current STRING API documentation for details.
enriched <- rba_string_enrichment(
ids = proteins_mapped,
species = 9606
)By default, results are split by category into a list of data frames.
We can inspect that structure with str():
str(enriched, max.level = 1)
#> List of 14
#> $ COMPARTMENTS :'data.frame': 25 obs. of 10 variables:
#> $ Component :'data.frame': 17 obs. of 10 variables:
#> $ DISEASES :'data.frame': 11 obs. of 10 variables:
#> $ Function :'data.frame': 12 obs. of 10 variables:
#> $ InterPro :'data.frame': 3 obs. of 10 variables:
#> $ KEGG :'data.frame': 45 obs. of 10 variables:
#> $ Keyword :'data.frame': 14 obs. of 10 variables:
#> $ NetworkNeighborAL:'data.frame': 5 obs. of 10 variables:
#> $ PMID :'data.frame': 100 obs. of 10 variables:
#> $ Process :'data.frame': 148 obs. of 10 variables:
#> $ RCTM :'data.frame': 59 obs. of 10 variables:
#> $ SMART :'data.frame': 1 obs. of 10 variables:
#> $ TISSUES :'data.frame': 12 obs. of 10 variables:
#> $ WikiPathways :'data.frame': 46 obs. of 10 variables:Let us see the “KEGG” results as an example. Below, we can see which terms of the KEGG pathways database were over-represented:
Please Note: Other services supported by rbioapi also provide Over-representation analysis tools. Please see the vignette article Do with rbioapi: Over-Representation (Enrichment) Analysis in R (link to the documentation site) for an in-depth review.
Functional enrichment plot
The enrichment-figure endpoint summarizes one enrichment category as
a plot. Use rba_string_enrichment_image() to control the
category, grouping, palette, ranking variable, output format, and number
of displayed terms. This example visualizes the KEGG enrichment for the
same protein set:
graph_enrich <- rba_string_enrichment_image(
ids = proteins_mapped,
species = 9606,
category = "KEGG",
image_format = "image",
save_image = FALSE,
group_by_similarity = 0.6
)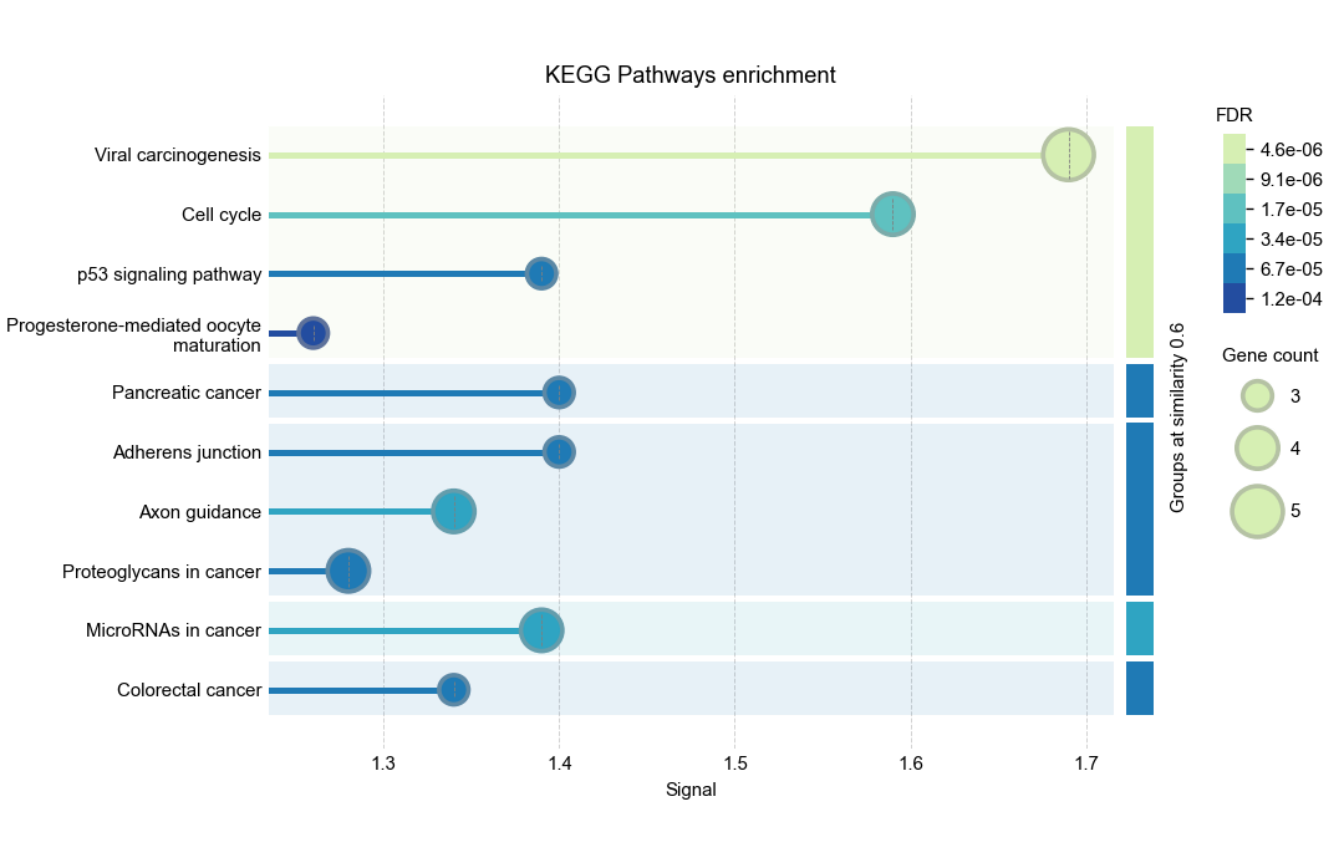
Visualization of enrichment analysis results
Protein-protein interaction enrichment
PPI enrichment tests whether the input network contains more interactions than expected for a similarly sized protein set drawn from the background proteome. It can therefore identify evidence of functional relatedness without relying on annotation enrichment.
rba_string_enrichment_ppi(
ids = proteins_mapped,
species = 9606
)
#> $number_of_nodes
#> [1] 14
#>
#> $number_of_edges
#> [1] 40
#>
#> $average_node_degree
#> [1] 5.71
#>
#> $local_clustering_coefficient
#> [1] 0.694
#>
#> $expected_number_of_edges
#> [1] 19
#>
#> $p_value
#> [1] 1.35e-05Get functional annotations
STRING maps proteins to multiple annotation resources. Use
rba_string_annotations() to retrieve all annotations
assigned to the input proteins rather than only the subset returned by
enrichment analysis. PubMed annotations are excluded by default because
they can make the response very large.
annotations <- rba_string_annotations(
ids = "9606.ENSP00000269305",
species = 9606
)
## Annotation responses can be large, so the result is not shown hereSet allow_pubmed = TRUE to include PubMed annotations
with the other categories, or set only_pubmed = TRUE to
retrieve only PubMed annotations. only_pubmed takes
precedence if both arguments are TRUE.
pubmed_annotations <- rba_string_annotations(
ids = "9606.ENSP00000269305",
species = 9606,
only_pubmed = TRUE
)See also in Functions’ manuals
Some rbioapi STRING functions were not covered in this vignette, please check their manuals:
How to cite
To cite STRING (Please see https://string-db.org/cgi/about?footer_active_subpage=references):
- Damian Szklarczyk, Rebecca Kirsch, Mikaela Koutrouli, Katerina Nastou, Farrokh Mehryary, Radja Hachilif, Annika L Gable, Tao Fang, Nadezhda T Doncheva, Sampo Pyysalo, Peer Bork, Lars J Jensen, Christian von Mering, The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest, Nucleic Acids Research, Volume 51, Issue D1, 6 January 2023, Pages D638–D646, https://doi.org/10.1093/nar/gkac1000
To cite rbioapi:
- Moosa Rezwani, Ali Akbar Pourfathollah, Farshid Noorbakhsh, rbioapi: user-friendly R interface to biologic web services’ API, Bioinformatics, Volume 38, Issue 10, 15 May 2022, Pages 2952–2953, https://doi.org/10.1093/bioinformatics/btac172
Session info
#> R version 4.6.1 (2026-06-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] rbioapi_0.8.3
#>
#> loaded via a namespace (and not attached):
#> [1] httr_1.4.8 cli_3.6.6 knitr_1.51 rlang_1.3.0
#> [5] xfun_0.60 otel_0.2.0 png_0.1-9 textshaping_1.0.5
#> [9] jsonlite_2.0.0 DT_0.34.0 htmltools_0.5.9 ragg_1.5.2
#> [13] sass_0.4.10 rmarkdown_2.31 grid_4.6.1 crosstalk_1.2.2
#> [17] evaluate_1.0.5 jquerylib_0.1.4 fastmap_1.2.0 yaml_2.3.12
#> [21] lifecycle_1.0.5 compiler_4.6.1 fs_2.1.0 htmlwidgets_1.6.4
#> [25] systemfonts_1.3.2 digest_0.6.39 R6_2.6.1 curl_7.1.0
#> [29] magrittr_2.0.5 bslib_0.11.0 tools_4.6.1 pkgdown_2.2.1
#> [33] cachem_1.1.0 desc_1.4.3